SOBRE A SÍNDROME DE ANGELMAN
GERAL
DEFINIÇÃO E HISTÓRICO
A síndrome de Angelman é uma disfunção neurológica rara que ocorre em aproximadamente um a cada quinze mil nascimentos (1 : 15.000). A estimativa é que exista mais de 12.000 pessoas com Angelman no Brasil.
A síndrome de Angelman caracteriza-se por:
- Atrasos no desenvolvimento neuropsicomotor
- Hipotonia
- Distúrbio do sono
- Refluxo
- Epilepsia
- Distúrbio de equilíbrio
- Estrabismo divergente
- Ataxia
- Apraxia global severa
- Dispraxia
- Deficiência intelectual
Em alguns casos a pessoa pode apresentar albinismo e é comum ter pele, olhos e cabelos mais claros do que o resto da família. Algumas pessoas com Angelman não andam. A grande maioria não fala. Eles não se tornam independentes e precisam de cuidado constante para o resto da vida.
Como se trata de uma síndrome rara, muitas pessoas e até mesmo médicos não tem conhecimento da síndrome de Angelman. Pode ser difícil entender de imediato todas as questões relacionadas à síndrome e você provavelmente terá várias perguntas a serem respondidas.
É sempre bom ressaltar também que a síndrome de Angelman é um espectro e nem todas as características se aplicam a todos os indivíduos. Cada pessoa é única e apresenta características e se desenvolve de maneira única.
Em breves termos, a síndrome de Angelman é causada por um acidente genético na região do cromossomo 15, todas envolvendo um único gene, o UBE3A.
A síndrome de Angelman foi identificada pela primeira vez em 1965 por um médico britânico, Dr Harry Angelman, daí a origem do nome da síndrome. O Dr. Angelman notou similaridades entre um pequeno grupo de crianças sem diagnóstico definido que estavam sob seus cuidados, indicando que elas tinham dificuldades semelhantes.
A princípio, muito pouco se sabia sobre a síndrome de Angelman. Em meados dos anos 80, avanços na medicina genética fizeram possíveis o aumento do número de diagnósticos aumentando, consequentemente, o número de pessoas diagnosticadas com a síndrome. O nome síndrome de Angelman foi adotado em 1982.
Em 2018 foi definida uma CID específica para Síndrome de Angelman. O código é o Q93.51,


UBE3A
ENTENDENDO O GENE UBE3A
Em todas as células do nosso corpo temos 22 pares de cromossomos e dois extra cromossomos que determinam nosso gênero (feminino são XX e masculino são XY). Para cada par, adquirimos uma cópia materna e outra paterna.
Sob o microscópio, cada cromossomo parece dois cilindros intercalados. Um cilindro é menor (denominado o braço “p”) e outro é maior (denominado o braço “q”). Cada braço possui diferentes regiões (as quais aparecem como bandas sob o microscópio) as quais são numeradas. Cada cromossomo abriga centenas de diferentes genes, pequenas porções de informação que juntos contribuem para formar cada pessoa de forma única.
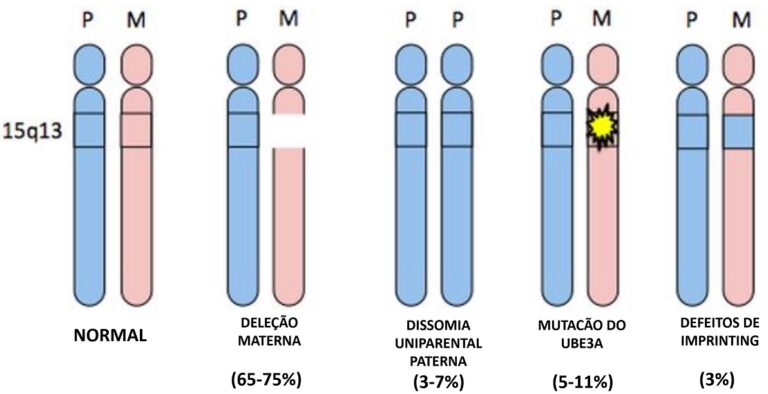
A síndrome de Angelman é causada por alterações num único gene, o UBE3A, situado no cromossomo 15, na região 11-13 do braço “q”, nomeada como 15q11-q13.
O gene UBE3A produz uma proteína chamada E6-AP, a qual é responsável pela manutenção das células. No cérebro, apenas a cópia materna deste gene está “ligada”. A cópia paterna está presente, mas “desligada”. Isto é denominado na genética como imprinting. Na grande maioria dos genes, quando ambas as cópias estão presentes e “ligadas” se uma das cópias se danificar, a outra cópia supre a necessidade, não havendo prejuízo. Porém, quando o gene é lido dessa forma, qualquer interrupção na ativação desse gene causa prejuízos.
A síndrome pode ser causada por 4 diferentes genótipos: deleção, mutação, dissomia uniparental e defeito de imprinting.
Em torno de 70% dos casos são causados por deleção, ou seja, perda de material genético na região q.11-13, onde está localizado o gene UBE3A. Cerca de 10% são causados por uma mutação no gene. Outros 5% a 10% são quando a pessoa recebe dois genes paternos, que estão silenciados, esses casos são conhecidos como dissomia uniparental (UPD). Os 5% a 10% restantes são casos de defeito de imprinting (ICD), isto é, apesar da cópia materna do UBE3A está perfeita, uma disruptura no centro de imprinting faz com que o gene seja silenciado.
Alguns casos de mutação e defeito de imprinting podem ser hereditários e, para famílias com diagnóstico desses genótipos, é necessário um aconselhamento genético.
Os sintomas da síndrome de Angelman aparecem geralmente aos seis meses de idade. Atrasos no desenvolvimento são geralmente os primeiros sinais. Não há, até o momento, nenhum tratamento de cura para a síndrome, mas há vários tipos de terapias que ajudam no desenvolvimento, como fisioterapia, fonoaudiologia, terapia ocupacional, etc, havendo também medicações para tratamento de eventuais crises convulsivas, refluxo, constipação e outras manifestações.